The biomedical words whose earliest recorded written instances are dated 1974 in the Oxford English Dictionary (OED) are listed in Table 1; as before, pharmacological words contribute the single largest group, and a few other items in the list can be antedated.
Table 1. Biomedical words (n=39) in the OED for which the earliest citations are from 1974 (out of a total of 272); I have found seven antedatings from 1 to 80 years
*Antedatings: monoclonality (1973); unifilarly (1963); [to] floss (1972); mechanosensory (1966); neurotrophism (1940); pathophysiologically (1972); photorefraction (1972)
†I have also found an instance of “unifilarly” from 1948, but meaning “[wound] with a single strand of wire, thread, or fibre”, not, as currently defined in the OED, “in a single strand of a DNA duplex”
Last week, discussing six medicines or groups of medicines that featured in the 1973 list, and for the invention of which Nobel Prizes in Physiology or Medicine were awarded, I highlighted two UK pharmacologists associated with them—James Black and John Vane—and discussed the latter. Now it is the turn of James W Black (1924–2010).
To be associated with research that leads to the development of a novel group of drugs is noteworthy. Black was associated with two. He won the Nobel prize for work that he did while working for two pharmaceutical companies, first ICI Pharmaceuticals and later Smith, Kline and French. He shared the prize with Gertrude B. Elion and George H. Hitchings (Figure 1). The citation read “for their discoveries of important principles for drug treatment”. Elion and Hitchings together developed methods of producing drugs that targeted the synthesis of cellular nucleic acids. This led, for example, to the development of purine antimetabolites, such as thioguanine and mercaptopurine for treating cancers, and allopurinol for treating gout. In contrast, Black chose to target plasma membrane-bound receptors. The method he used was to take an agonist at a receptor and modify its structure to produce an antagonist.

Figure 1. The winners of the 1988 Nobel Prize for Physiology or Medicine; from left to right James W Black, Gertrude B Elion, George H Hitchings
In 1948 Raymond P Ahlquist, searching for a chemical substitute for the adrenoceptor agonist ephedrine, discovered different properties of the compounds he synthesized and consequently classified adrenoceptors into two subtypes, α and β. His paper has been cited over 4500 times. The adrenoceptors were later further subdivided, e.g. the β receptors into β1, β2, and others. In 1957, Irwin Slater of Eli Lilly discovered that dichloroisoprenaline was an adrenoceptor antagonist. Black looked for beta-adrenoceptor antagonists by modifying the structure of the agonist isoprenaline, and came up with pronethalol. Because of adverse effects pronethalol was replaced by propranolol; other drugs followed (Table 2).
Table 2. First-in-class and me-too beta-adrenoceptor antagonists (beta-blockers) and their innovative features
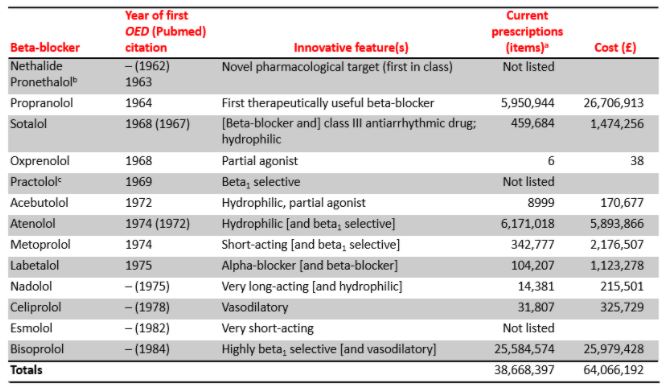
aNumbers of generic items prescribed by GPs in NHS England from September 2019 to August 2020 (source openprescribing.net)
bOriginally called nethalide; renamed pronethalol in 1963
cFailed owing to adverse reactions
Black’s invention of the histamine H2 receptor antagonists was based on a similar process. In the 1940s Bernard Halpern showed that although conventional antihistamines protected guinea-pigs against the adverse effects of histamine, the animals died from gastric perforation. Then in 1953, in a paper that has been cited nearly 900 times, Drew Kay, who was professor of surgery in the Western Infirmary in Glasgow when I was a medical student, showed that maximal doses of histamine could be used to stimulate gastric acid production, provided the non-gastric effects of the histamine were first blocked by a conventional antihistamine such as mepyramine. In elegant dose-response experiments he showed that 4 mg of histamine per 10 kg body weight produced maximal inhibition of gastric acid secretion in all the subjects he studied. Kay’s “augmented histamine test” then became a standard method of studying gastric acid secretion, for example to establish the completeness of vagotomy after surgical treatment of peptic ulceration.
In 1951, Grossmann et al. discovered three compounds that inhibited histamine-stimulated gastric acid secretion in dogs, although two of them did so only after first stimulating it; the third was a triazole derivative. In 1966 Ash & Schild proposed that the non-gastric histamine receptors should be called H1 and in 1972 Black et al. proposed that the gastric receptors be called H2. By that time he and his colleagues had screened over 200 histamine analogues as potential H2 antagonists and found one, guanylhistamine, a partial agonist. This was followed by burimamide, an imidazole derivative, which would have appeared in the 1972 list, had there been an entry for it in the OED. Burimamide could not be given orally, and although its successor, metiamide, was orally active it caused granulocytopenia. However, the next compound in line, cimetidine (which will feature in next week’s list), hit the mark (Table 3).
Table 3, First-in-class and me-too histamine H2 receptor antagonists and their innovative features and disadvantages
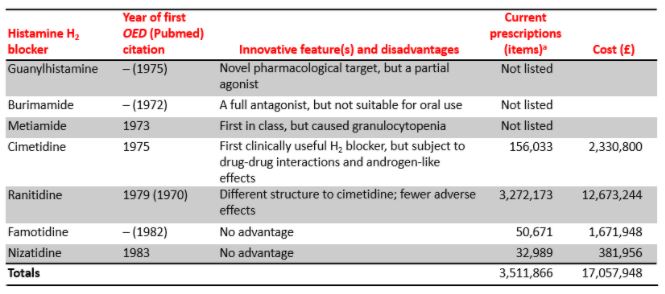
aNumbers of generic items prescribed by GPs in NHS England from September 2019 to August 2020 (source openprescribing.net)
Other H2 blockers not listed in the OED and not in general clinical use include tiotidine (1979), oxmetidine (1980), etintidine (1982), lamtidine (1983), loxtidine (1983), icotidine (1984), lupitidine (1986), and ebrotidine (1992)
These inventions, the beta-blockers and the histamine blockers, transformed the management of cardiovascular disease and peptic ulceration. Black’s prize was well deserved.
Jeffrey Aronson is a clinical pharmacologist, working in the Centre for Evidence Based Medicine in Oxford’s Nuffield Department of Primary Care Health Sciences. He is also president emeritus of the British Pharmacological Society.
Competing interests: None declared.
