By Hugh Davies
The Food and Drug Administration’s (FDA) initiative
Over the last three decades seeking consent of patients or volunteers to join a medical research study has become dominated by an increasingly lengthy, opaque and Participant Information Sheet (PIS), shielding the researcher from liability. The FDA argues that
- Informed consent documents are often long, complex, and legalistic
- The informed consent process does not take full advantage of appropriate innovations (e.g., images, videos, technology) that can facilitate understanding
- More work is needed to fulfil the promise of truly participant-centred and participant-partnered informed consent
To address these issues, they have proposed revision of the content, organization, and presentation of information to facilitate a prospective subject’s decision about whether to participate in the research (Key Information and Facilitating Understanding in Informed Consent ) holding a virtual public webinar entitled “Informed Consent – More than Just Another Document to Sign?” in November 2024. Their central proposal would be a requirement that consent must
“begin with a concise and focused presentation of the key information that is most likely to assist a prospective subject or legally authorized representative in understanding the reasons why one might or might not want to participate in the research”
They would propose that this should be seen as a “key fact summary”. As such it wouldn’t provide all details, rather it would be an introduction, a template for the conversation and a map with links to further details in sections providing mandatory and additional information. The following content was suggested:-
- Voluntary Participation and Right to Discontinue Participation
- Purpose of the Research, Expected Duration, and Procedures to Be Followed
- Reasonably Foreseeable Risks and Discomforts
- Reasonably Expected Benefits
- Appropriate Alternative Procedures
- Compensation and Medical Treatments for Research-Related Injuries
- Costs Related to Subject Participation
When developing this, the FDA would encourage researchers to meaningfully involve patients and patient groups to seek their views on what would likely be key information from their perspective, further promoting patient involvement in research from a very early stage.
Why is this initiative important?
While these requirements would only apply to research regulated by the FDA, its influence stretches much further afield and these are in line with other jurisdictions (EU Horizon 2020, UK Health Research Authority), current ethical thought (Flory and Emanuel 2004, Nishimura et al 2013, Fons-Martinez J et al) and medical decision making theory (Gillies et al, Ottawa Decision Support Framework)
However the FDA recognises thisWorkouts is just a start and much would rest on the shoulders of researchers. One size would not fit all and it encourages researchers to look for ways to improve consent. The recommendations from the Clinical Trials Transformation Initiative complements the FDA’s work and could be a start:
“1. The informed consent process should involve an ongoing, interactive conversation between the research participant and the research staff, beginning with initial consideration of study participation and continuing through study completion.
- The informed consent process should be customized to meet the particular needs of individual study participants.
- The person or persons obtaining consent should be skilled in communicating trial-specific information and be responsive to the needs and concerns of individual research participants.
- A discussion tool, not intended as a required regulatory compliance document could be used as part of the consent process to ensure the following:
- The specific needs of each study participant are considered,
- Key elements of the trial are reviewed and addressed,
- Interactive techniques are used to facilitate participant understanding of the information imparted.
- This tool could be used for documenting the informed consent process.
- Study participants should be provided with available resources to enhance their understanding of clinical trials, including sample questions to ask the investigator so he/she can better engage in a dialogue about the benefits and risks of participation.
- The informed consent document should be viewed as supportive to the consenting process, rather than the primary focus.”
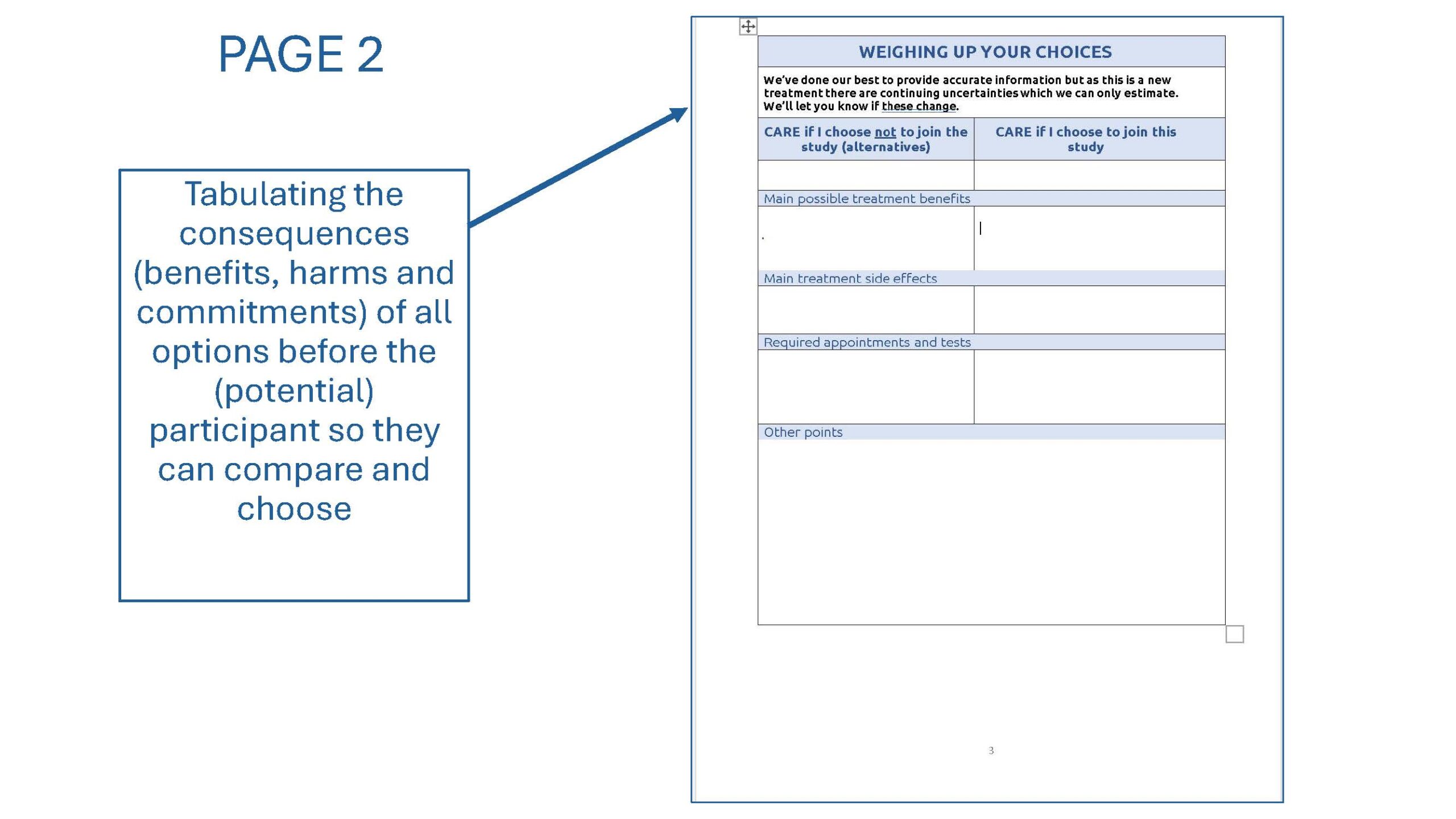
On the Oxford ‘A’ Research Ethics Committee (REC), we met similar problems. Current research proposals mostly provide adequate detail (even if not in an easily comprehensible format), but often fail to support decision making, the central purpose of consent. To address this we proposed an “Information and Decision Aid” :
- Page 1 This is headed by a simple, comprehensible title, then an invitation, explanation that participation is the patient’s choice and a table of “key facts” determined during research design in collaboration with patient groups (PPI).

- Page 2 This would be a table laying out the benefits and harms of both joining an not joining the study along the lines recommended by Svobodova et al.
- Page 3 would be an easy-to-read schematic diagram outlining what taking part in the study would involve.

- Page 4 Provides space to personalise consent by allowing the participants to put down (and seek answers to) their concerns and uncertainties and then document how understanding was confirmed aligning it with medical decision making theory.

We proposed that this could be used as an introduction to the study, sent in advance, a template for the conversation between the researcher and possible participant, an introduction / summary at the top of the Participant Information Sheet or a check when consent is signed off, alongside the Informed Consent Form.
Conclusion
If the process of informed consent is to be fit for purpose and ensure any potential participant has been helped to make a decision they can be happy with, the current FDA’ s initiative provides us with a springboard which will align guidance, ethical and decision making theory. It is currently only draft guidance and, given the current political climate, there will be uncertainty as to its final fate but we would be remiss not to take up these ideas and the opportunity now presented to us.
Author: Hugh Davies (with thanks to the members of the Oxford “A” REC for their contributions)
Affiliation: Chair, Oxford “A” REC
Competing Interests: None to declare