The European Medicines Agency (EMA) has issued its long anticipated new policy (policy 0070) on prospective access to clinical trial data, and is now in consultations to figure out the details of its implementation. We were invited to join these ongoing consultations, and have previously reported on the debate here and here.
We have been particularly concerned about the anonymization and redactions of the content of clinical study reports (CSRs), and especially concerned about the approach proposed by some in industry.
But now we are getting really worried. Current drafts of the EMA’s evolving guidance documents for the anonymization of CSRs leave too much leeway for creative interpretation of acceptable anonymization practices, and an EMA follow-up meeting on 7 September made clear that some industry associations are pressing to apply a standard known as the TransCelerate approach. While almost all approaches sound reasonable (after all, they are intended to protect the anonymity of trial participants—a good thing), the TransCelerate redaction approach would cripple the usefulness of CSRs.
Take a look for yourselves. Figure 1 (below) is a page from a Tamiflu CSR (Research Report No. 1005291) that Roche released to us after a four year long battle for access. Figure 2 shows what would be likely to happen if Roche applied the TransCelerate redaction standard to that same document. Applying the TransCelerate approach takes the Tamiflu document and turns it into a page of black boxes. For instance, all dates relating to individual trial participants have to be redacted, as well as other patient information such as sex, age, weight, height, race, ethnicity, and socioeconomic information. All patient narratives would also have to be removed.
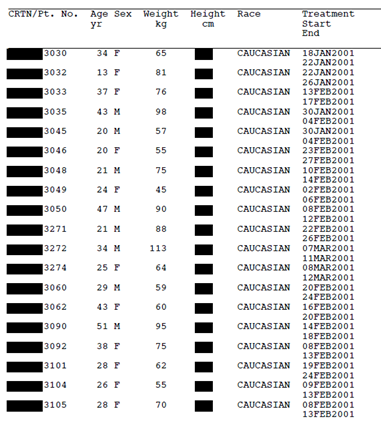
Figure 1: Line listing from Tamiflu trial WV16277 (Research Report No. 1005291) redacted by Roche for public release. Available from http://dx.doi.org/10.5061/dryad.77471

Figure 2: Line listing from Tamiflu trial WV16277 (Research Report No. 1005291) redacted according to the TransCelerate guidance. Available from http://dx.doi.org/10.5061/dryad.77471
Why “likely to happen” and not “happen”? Because we had to create figure 2 ourselves. Ideally, those advocating a redaction approach would send around shared examples for the rest of us to see and discuss. But there were no clear examples at the EMA meetings.
Using redactions to assure the anonymization of data in CSRs is emerging as a make or break issue for the success of the EMA initiative. The intensive redaction of the TransCelerate approach risks nullifying most of the progress towards transparency made so far in Europe.
The rationale for policy 0070 is the public posting of documents used by the EMA to make a decision on a market authorization application for pharmaceuticals or biologicals—to allow maximum transparency in decisions. Anyone will be able to access the documents after a simple registration procedure, and they should be able to reconstruct the trial or evidence development programme and read the trial protocol, its amendments, the statistical analysis plan (and any of its amendments), and a core report (an unabridged reporting of the trial, usually hundreds of pages long)—see here for a background paper on CSRs.
This sounds a lot simpler than it is, as CSRs can be immensely complicated documents and it is likely that only researchers and industry will read them in detail. Regardless of who reads the documents and why, the publication of regulatory documents makes sense only if they are clear and their content is understandable, verifiable, and has not been censored or shortened to such an extent that the facts reported are not compromised.
This absolute requirement for clarity generates a tension, which threatens to dilute the EMA’s revolutionary transparency initiative. With its strong emphasis on maximizing data utility, the EMA is trying to keep redactions and manipulation of text to a minimum. Yet, at the same time, some parts of industry are proposing to use methods, such as TransCelerate, to further anonymize the documents to prevent re-identification of participants (and even redacting the names of trial investigators and supervisors, when some of that information could be readily obtained from public registries).
The current draft guidance by the EMA (which we have seen, but the EMA has requested it not be publicly circulated) has deemed two approaches to data anonymization as acceptable:
• Common sense approaches or rules of thumb
• Quantitative methods that analyze the data itself to measure the risk of re-identification
The second method applies a more quantitative approach to deciding how much information to redact or modify in a CSR. This generally ensures that minimal manipulations are performed to the documents and maximizes their utility. The first option is more subjective, and would allow data custodians to apply extensive redactions under any “rules of thumb”—which is illustrated by the example in figure 2. To avoid this, any rules still need to be based on a quantitative analysis to justify them—there needs to be defensible rigour in coming up with anonymization rules. Unsubstantiated “common sense” could mean anything. It is important that this rigour be a clear expectation in the EMA’s anonymization guidance as well.
The vagueness of the EMA draft guidance, while understandable in such a consultation and development phase, nonetheless leaves the door open to the severe redaction of CSRs. Our practical experience has shown the value of serious adverse event narratives and line listings for the appraisal of a trial and the replication of its results in the context of systematic reviews.
It is time to get down to examples. Perhaps the next step should be the dissemination of a demonstration CSR and dataset to enable data anonymization using the different methods. This would allow the public to gauge their impact on clarity and eventual utility.
Khaled El Emam is the Canada research chair in electronic health information at the University of Ottawa, an associate professor in the department of pediatrics, and is cross-appointed to the school of electrical engineering and computer science.
Tom Jefferson, reviewer, Cochrane Acute Respiratory Infections Group, 00187, Roma, Italy.
Peter Doshi, associate editor, The BMJ.
Competing interests: TJ and PD were co-recipients of a UK National Institute for Health Research grant (HTA – 10/80/01 Update and amalgamation of two Cochrane Reviews: neuraminidase inhibitors for preventing and treating influenza in healthy adults and children—http://www.nets.nihr.ac.uk/projects/hta/108001).
TJ receives royalties from his books published by Blackwells and Il Pensiero Scientifico Editore, Rome. TJ is occasionally interviewed by market research companies for anonymous interviews about Phase 1 or 2 pharmaceutical products. In 2011-2013, TJ acted as an expert witness in a litigation case related to oseltamivir phosphate; Tamiflu [Roche] and in a labour case on influenza vaccines in healthcare workers in Canada. In 1997-99 TJ acted as a consultant for Roche, in 2001-2 for GSK, and in 2003 for Sanofi-Synthelabo for pleconaril (an anti-rhinoviral, which did not get approval from the Food and Drug Administration). TJ was a consultant for IMS Health in 2013, and in 2014 was retained as a scientific adviser to a legal team acting on the drug Tamiflu (oseltamivir, Roche). In 2014-15, TJ was a member of two advisory boards for Boerhinger and is in receipt of a Cochrane Methods Innovations Fund grant to develop guidance on the use of regulatory data in Cochrane reviews. TJ has a potential financial conflict of interest in the investigation of the drug oseltamivir. TJ is acting as an expert witness in a legal case involving the drug oseltamivir (Roche). TJ is a member of an Independent Data Monitoring Committee for a Sanofi Pasteur clinical trial.
PD received €1500 from the European Respiratory Society in support of his travel to the society’s September 2012 annual congress in Vienna, where he gave an invited talk on oseltamivir. PD gratefully acknowledges the American Association of Colleges of Pharmacy for its funding support ($10 000) for a study to analyze written medical information regarding the possible harms of statins. AACP had no involvement in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of this manuscript.
Competing Interest: KEE has financial interests (including equity) in Privacy Analytics Inc., a University of Ottawa and Children’s Hospital of Eastern Ontario spin-off company, which develops anonymization software for the health sector. He also receives royalties on his published books from Francis & Taylor, O’Reilly Media, Wiley, and Trafford.