The critical involvement of central pathways in the spread of amyotrophic lateral sclerosis (ALS) is well-recognised both clinically and neuropathologically, and post-mortem studies have classified distinct pathological disease stages. Recently, neuroimaging studies have proposed four main anatomical stages of disease spread: the first stage involving the corticospinal tract (CST), stage 2 involving corticorubral and corticopontine tracts, stage 3 encompassing corticostriatal pathways and final involvement of preforant path marking stage 4.
In their recent article published in JNNP, Kassubek and colleagues use a diffusion tensor imaging (DTI) approach to longitudinally explore these anatomical pathways with respect to disease staging and spread, using a specific tract of interest (TOI)-based technique to also target the analysis to the individual patient. Their overall findings support longitudinal change along these four main anatomical pathways, particularly stages 1 to 3 (figure B: Significant longitudinal alterations and ALS related tract structures, calculated by whole-brain-based spatial statistics of 67 patients with ALS vs 31 controls). The progressive structural dysfunction (seen as a change in functional anisotropy, FA) also correlated with clinical functional scores and disease duration across all tract systems, suggesting the potential of this technique to be used as a non-invasive prognostic biomarker.
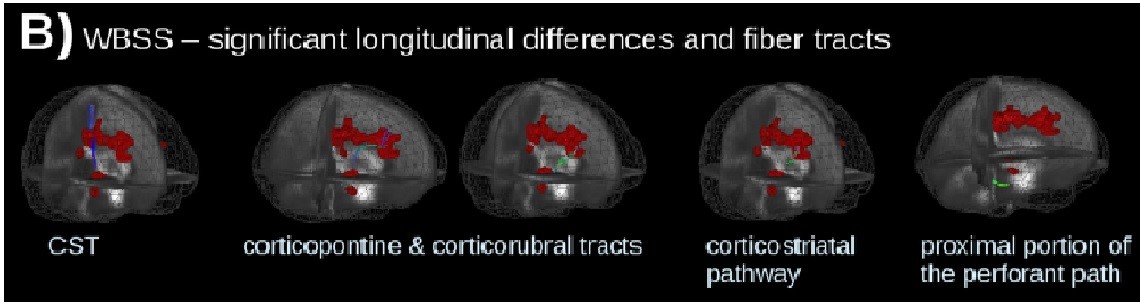
This study complements current understanding of disease progression and patterns of spread in ALS, highlighting several important points for consideration. Firstly, the cross-sectional analysis reaffirms significant CST changes, supporting the primacy of the motor pathway in disease manifestation. White matter involvement in other areas, mainly the frontal, brainstem and hippocampal regions, also correlates well with the four neuropathological disease stages described in separate post-mortem ALS studies, suggesting that DTI may also be able to assess disease staging in vivo. Of particular interest, the authors’ use of TOI techniques to map and track axonal loss over time at the level of the individual patient is particularly relevant, as methodological variability in previous DTI studies have cast significant doubt regarding suitability of this technique for individual patient analysis. Given the heterogeneity of ALS, an ability to individualise analysis would clarify intrinsic differences between subgroups, enable more suitable patient stratification for clinical trials and guide personalised therapeutic approaches.
From this point, ongoing characterisation of central disease patterns in much larger cohorts will enable the eventual translation of traditional histopathological and molecular findings of ALS into a dynamic clinical context. Overall, it remains hopeful that an integrated multimodal neuroimaging approach including DTI, combined with clinical and neurophysiological applications, will lend the ability to develop individualised ‘brain maps’ to identify and explore in vivo trajectories of disease in an integrated fashion, stratifying more effectively for clinical trials and for potential use as a marker of treatment efficacy.