In the current issue of JNNP, Watanabe and colleagues published a genome-wide association study (GWAS) in ALS to explore the effects of genetic variants in the disease course of sporadic ALS patients.
ALS is an incurable neurodegenerative disease that affects the motor neurons in the cortex, brainstem, and spinal cord, typically resulting in death within 2 – 3 years. Even though enormous advances in the comprehension of the disease have been achieved during the last decade, the huge clinical and genetic heterogeneity of the disease have hindered the development of new disease-modifying treatments. With the aim to explore the heterogeneity across ALS spectrum, a number of GWAS using large-scale genotyping of single nucleotide polymorphisms (SNPs) have failed to generate consistent results and reported associations were not strong enough to develop suitable disease models.
In the study, Watanabe and colleagues explore the impact of genetic variants (SNPs) in different patterns of functional decline in patients with sporadic ALS. A total of 465 ALS patients were clustered according to the longitudinal functional score (ALSFRS-R) in four groups: a rapid decline cluster, an intermediate decline cluster, a sigmoidal decline cluster and a moderate decline cluster. Amongst the 572.983 SNPs studied using genome-wide analysis, seven SNPs were associated with the rapid decline cluster (odds ratio: 5.5 to 5.84). Specifically, homozygosity for the minor alleles of the seven SNPs (linkage disequilibrium block) was associated with decreased expression of TNN (the gene that encodes Titin, a sarcomere protein). Finally, the authors described down-regulation of Titin in immortalised lymphocytes lines from such patients with ALS.
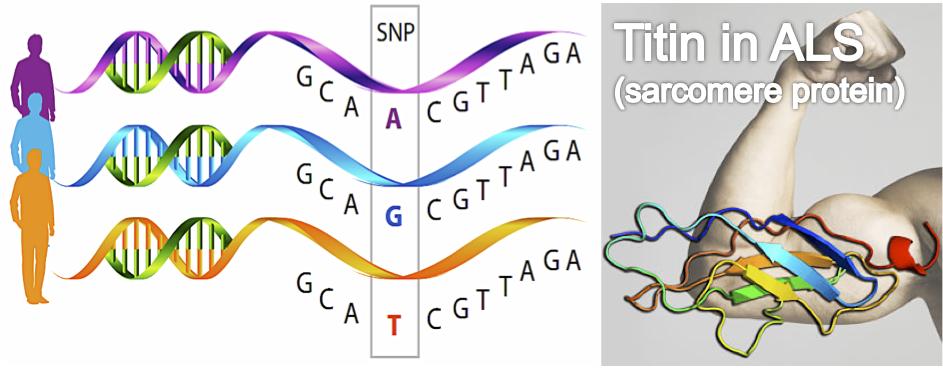
The studying of the genetic factors that influence the clinical course of ALS is extremely valuable in the design of new clinical trials. This study is the first that identifies genetic factors associated with rapid functional decline in sporadic ALS patients. The seven SNPs related to functional decline were associated to decreased Titin expression, an essential sarcomere protein. These results may suggest that Titin determines ALS disease progression through its role in muscle function. Previous studies have related Titin with different myopathies (e.g. limb-girdle muscle dystrophy). Given that there are not previous studies of Titin in ALS, these results represent a new opportunity to explore the role of Titin in ALS pathogenesis. From a clinical point of view, the impact of Tinin on the clinical disease course must be confirmed in future prospective studies, which must explore these genetic variants in ALS patients with different genetic backgrounds.
Finally, it is clear that this study provides a new candidate that might help to understand the complex heterogeneity behind the ALS spectrum.
Read more at http://jnnp.bmj.com/content/87/8/851.full.pdf