Sporadic Creutzfeldt-Jacob disease (sCJD) is a complex neurodegenerative disorder characterized by the accumulation of misfolded human prion protein (PrPSc). Classically, sCJD presents with neurological symptoms and signs of central nervous system (CNS) dysfunction, including cognitive impairment, cerebellar dysfunction, myoclonus, visual impairment and pyramidal signs, amongst others. Moreover, clinical diagnostic criteria requires the presence of specific brain MRI and EEG changes suggestive of the disease. Interestingly however, recent novel technology detecting misfolded prion protein in the peripheral nervous system (PNS) has now also been described.
In the April issue of JNNP, Baiardi and colleagues explored the frequency of peripheral neuropathy in patients with sCJD. Medical records were retrospectively reviewed from more than 300 sCJD patients, collecting both clinical features and neurophysiological data. Patients were classified into major clinical–pathological subtypes using the genotype at the polymorphic codon 129 (methionine, M; or valine, V) in the prion protein gene (PRNP) and the type (1 or 2) of abnormal prion protein (PrPSc) accumulating in brain tissue. In addition, western blotting and real-time quaking-induced conversion assay (RT-QuIC) were performed in postmortem peripheral nerve tissue (sural) samples from 14 patients. In summary, the frequency of symptoms/signs suggestive of PNS compromise was higher in the VV2 (39.3%) and MV2K (44.6%) patients than in the MM (V)1 group (9.1%). From the 40 VV2-MV2K patients that had undertaken nerve conduction studies and electromyography, 13 had signs of polyneuropathy. Finally, prion RT-QuIC was positive in all nerve samples, while western blotting was positive in just two samples (figure 1).
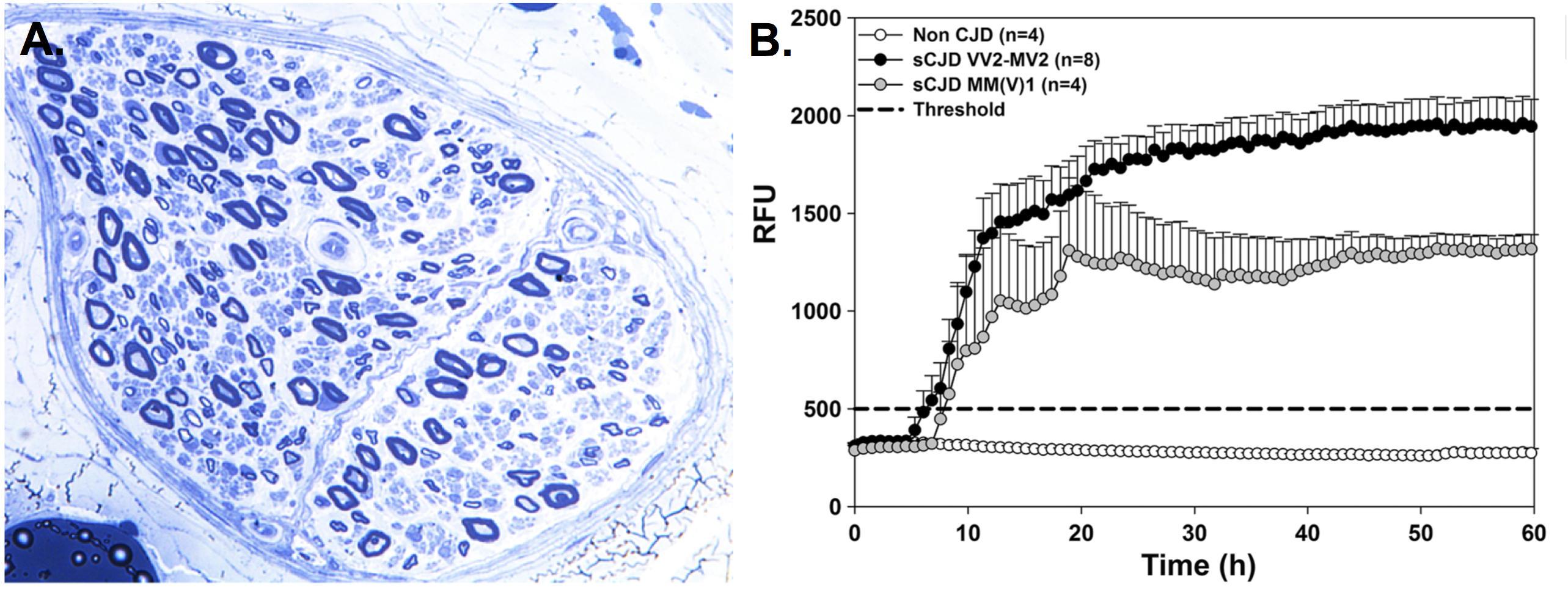
This is the first study that explores the compromise of the PNS in a large cohort of sCJD patients, revealing peripheral symptoms/signs in 40% of patients with VV2 and MV2K. Of interest, the presence of PNS dysfunction seems dependent on the prion-strain, with a lower frequency of dysfunction found in patients with the typical MM(V)1 subtype. Even though the presence of PrPSc has been described previously in peripheral nerve tissue, the present study detected the presence of PrPSc in all sural nerve samples explored. This article thus suggests that CJD should be considered in the differential diagnosis of patients with neuropathy when accompanied with initial symptoms/signs of CNS dysfunction, and as such, provides valuable insights into sCJD heterogeneity.
Read more at https://jnnp.bmj.com/content/90/4/424