For second #GUTBlog, Professor El-Omar has invited Dr Kevin Monahan, Consultant Gastroenterologist at St Mark’s Hospital, London, who is the first author of the “Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG)” published in Gut.

We know that approximately 35% of colorectal cancer (CRC) risk is due to heritable factors, and that almost 30% of the population in the United Kingdom (UK) have a family history of CRC. The optimal management of people at high hereditary risk of CRC is known to improve survival after a diagnosis of cancer, and understanding this risk in population also provides many opportunities for cancer prevention.
In Gut [1], we present our guideline for the multidisciplinary lifelong care of people with hereditary risk factors for colorectal cancer (CRC) . The scope of this guideline includes diagnostic, endoscopic, surgical management as well as other preventative strategies, focused on the reduction in lifetime CRC risk in the high risk population.
Conversely there is a proportion of the UK population which has relatively low genetic CRC risk. In addition, because CRC is a common disease, a family history of CRC may be expected in many people who may not necessarily have a significantly increased risk of CRC themselves.

Therefore hereditary CRC risk is potentially relevant to a large proportion of the population, and there may be much to be gained by identifying who is truly at increased risk, and offering more effective interventions to these people.
How might we effectively offer the ‘right tests’ to the ‘right people’? The first step is to understand which people are at high risk is by offering a molecular assessment of possible genetic risk, combined with a systematic assessment of their personal and family history of colorectal cancer and polyps.
We sought to identify groups of people who are likely to gain from better access to genetic testing and molecular assessment of tumours in families with a history of CRC. These include people with a family history of CRC or genetic syndromes, and also people with a personal history of multiple polyps or CRC, especially those with early-onset CRC.
By offering a ‘personalised-medicine’ approach, investigations and treatments may be tailored to people according to their level of risk. For example, a person with an estimated increased CRC risk related to a family history of CRC may have this risk significantly mitigated by colonoscopy, delivered with sufficient quality assurance. However accurate risk assessment based on understanding an individual’s genetic or molecular risk factors offers specific advantages in prevention and treatment – for example in our recommendations we link molecular assessment of risk to the timing and intensity of endoscopic follow-up. In the near future polygenic risk scores may determine the application of CRC preventative interventions in the whole population including national screening.
Inherited CRC syndromes with a very high lifetime CRC risk may result from specific genetic mutations in cancer predisposition genes (now often called ‘pathogenic variants’). This CRC risk approaches 100% for people with mutations in the APC gene causing familial adenomatous polyposis (FAP), a very rare disease. Lynch Syndrome, on the other hand, is a common disease which affects between 1 in 100 and 1 in 400 people. However it is under-recognised and in the UK we only know of about 5-10% of people with this condition. Because universal testing enhances recognition and diagnosis, we have recommended testing for Lynch Syndrome in all new cases of colorectal cancer in the UK, consistent with guidelines previously published by NICE for England [2].
We have also developed gene-specific guidelines for people with Lynch Syndrome, a condition defined by the diagnosis of mutations in the mismatch repair (MMR) genes. Lifetime CRC risk varies between 15% and 70%, depending on the specific faulty underlying mismatch repair gene. Thus management of Lynch Syndrome should vary according to which gene is implicated, and there is a suggestion that Lynch Syndrome may actually represent four different syndromes related to specific underlying MMR gene defects.
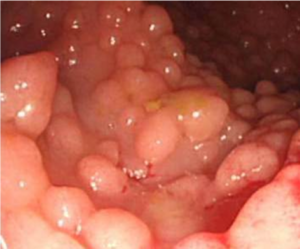
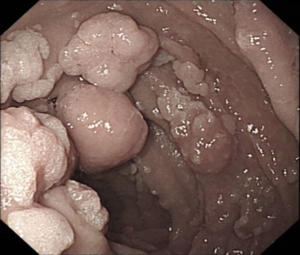
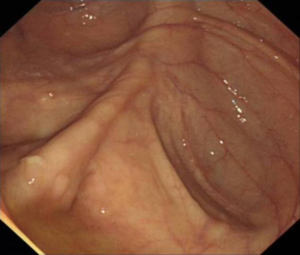
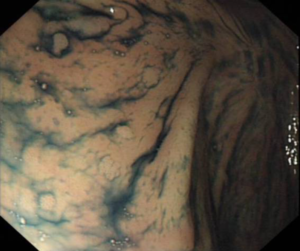
Surgical approaches following a diagnosis of CRC may differ in people with hereditary CRC syndromes factors compared to the general population. Colectomy may prevent CRC in people with some genetic syndromes, but preventative resection should be employed in different ways in people with FAP or Lynch Syndrome.
Surgery is usually offered to people with FAP in their teenage years as primary prophylaxis, although decision making about the timing and extent of resection may be related to an individual’s APC genotype, family history of desmoid disease, and other factors. In those with Lynch Syndrome and a new diagnosis of colorectal cancer, extended colorectal resection may be considered for secondary prophylaxis, but only when gene-specific and other clinical factors are carefully weighed-up
The lifelong care for people at hereditary CRC risk also includes lifestyle advice, as even the most potent of genetic risk factors may be modified by factors such as obesity, smoking and diet. There is also evolving evidence about how we might use medication to reduce risk, for example aspirin in people with Lynch Syndrome which we have recommended. However in people with polyposis syndromes the benefit of non-steroidal anti-inflammatories (NSAIDs) is less clear despite the publication of earlier initially promising evidence.
We have outlined the service and structures required to ensure the lifelong care of our patients. We suggest the nomination of people within local multidisciplinary CRC teams (‘genomic champions’) who are specifically responsible for ensuring pathways from diagnosis through to treatment and follow-up for people at hereditary CRC risk [3].
The optimal care as defined in this guideline requires development of local and regional networks of clinicians managing hereditary risk in primary, secondary and tertiary care. In secondary care many institutions operate family history clinics which facilitate patient-centred quality-improvement, however this should be prioritised to reduce regional variability in provision. Improved access to genomic testing is being facilitated nationally by mainstreaming projects. However in order for clinicians to develop relevant experience of this testing, a structured support network is required linked to tertiary care and the genomic laboratory hubs. We believe this is a cornerstone which will help to reduce variation in access to diagnosis, prevention and treatment of people at hereditary CRC risk.”
References
1 Monahan KJ, Bradshaw N, Dolwani S, et al.Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2019;69:411–44. doi:10.1136/gutjnl-2019-319915
2 Excellence TNI for H and C. Molecular testing strategies for Lynch syndrome in people with colorectal cancer (NICE Diagnostics Guidance DG27)<br>. Diagn Guid. 2017;27:1–37.https://www.nice.org.uk/guidance/dg27 (accessed 2 Nov 2019).
3 Monahan KJ, Alsina D, Bach S, et al.Urgent improvements needed to diagnose and manage Lynch syndrome. 2017;356. doi:10.1136/bmj.j1388